
Contributors
Fabio Valente a, Johann Joets a, Alain Charcosset a, Laurence Moreau a, and Shawn Yarnes b
Institut National de la Recherche Agronomique (INRA) a, The Integrated Breeding Platform b
Summary
Use OptiMAS to select and cross the best of 297 genotyped individuals for the next round of marker-assisted recurrent selection (MARS). Calculate the predicted genetic value of genotyped individuals based on 11 target QTL. Design crosses based on the likelihood of combining all favorable alleles into a single individual.
- Introduction
- Demonstration Data
- Run OptiMAS
- Prediction
- Selection
- Intermating
Introduction
OptiMAS (Blanc et al. 2013) predicts crossing strategies that optimize the likelihood of assembling favourable alleles into a target genotype. Molecular markers in the vicinity of favourable parental QTL are used to trace the movement of QTL between generations. OptiMAS uses genotype data to predict the probability of allele transmission in different marker-assisted selection (MAS) schemes and mating designs (intercrossing, selfing, backcrossing, double haploids, RIL), allowing some generations to be considered without the need to genotype. Selection and crossing strategies are based on expected progeny genotypes. OptiMAS supports decision-making associated with the marker-assisted breeding plan generated by the Molecular Breeding Planner.
OptiMAS is a Breeding Management System standalone tool that can be launched from the workbench or from a desktop icon. OptiMAS has a graphical user interface, but can also operate from command line. For more information on command line operation and additional demonstration examples see the OptiMAS development site or read the full manual (pdf). ?
Demonstration Data
The maize (Zea mays) data set comes from a mulitparental population described by Blanc et al. 2006 and 2008. Eleven QTL were detected for silking date based on six connected F2 populations, based on 150 individuals each. The F2 populations were obtained from a half-diallel design between four unrelated maize inbred lines (DE, F283, F810 and F9005). A set of 34 markers was selected with at least three microsatellite markers following each QTL. Two cycles of marker-assisted recurrent selection (MARS) were performed with a step of selfing occuring before each intermating. In this example, OptiMAS is used at the last cycle to select the best individuals among 397 genotyped plants for the next cycle of MARS.
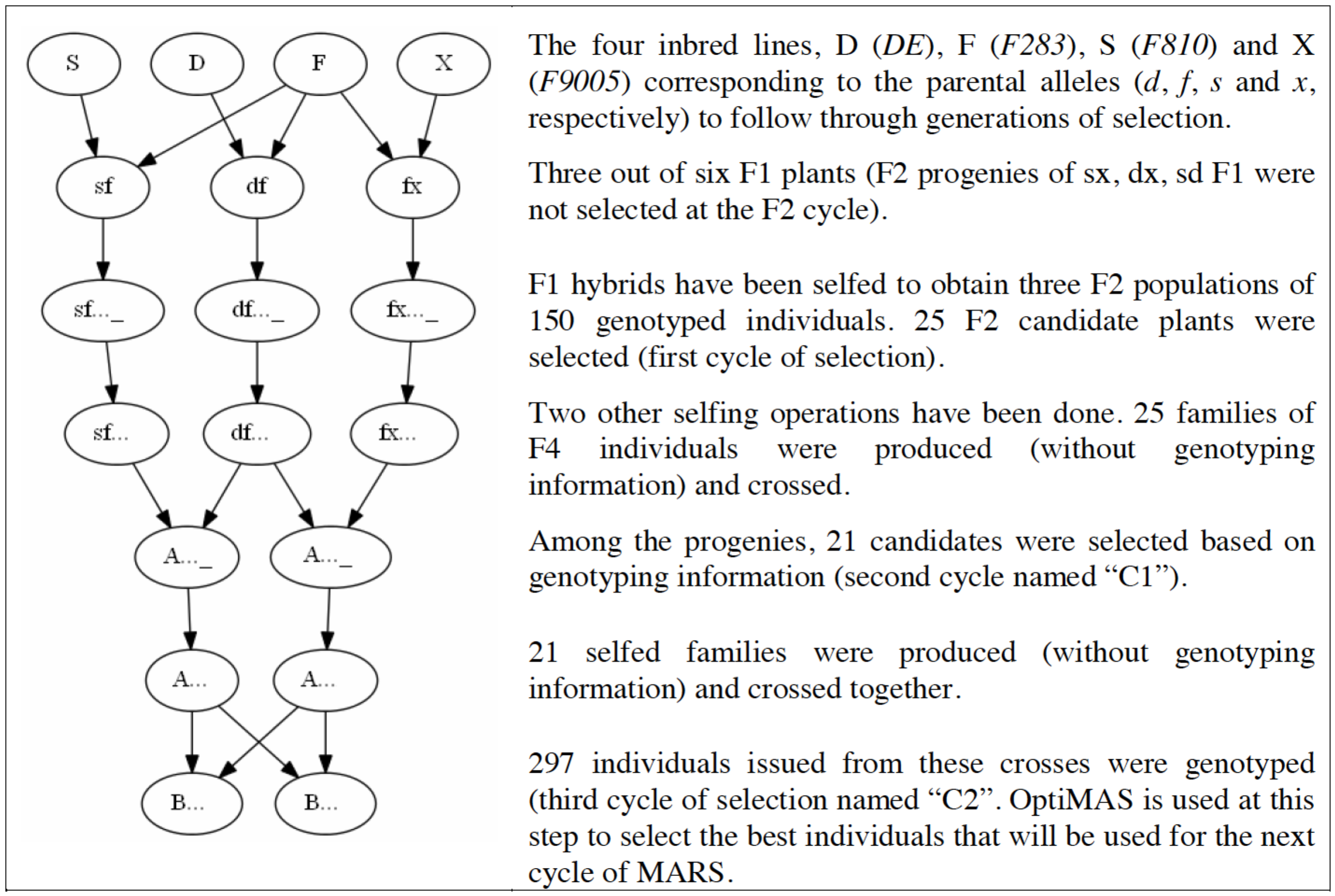
MARS Breeding Scheme (Reprinted with permission from Blanc et al. 2006, 2008)
Genetic Map File (.map)
The map file is supplied by the user and specifies QTL of interest, flanking makers, and favourable alleles. QTL identified by the Breeding Management Systems Breeding View application, and other external applications, can be used to create the genetic map file for OptiMAS.
Example File: ?blanc.map
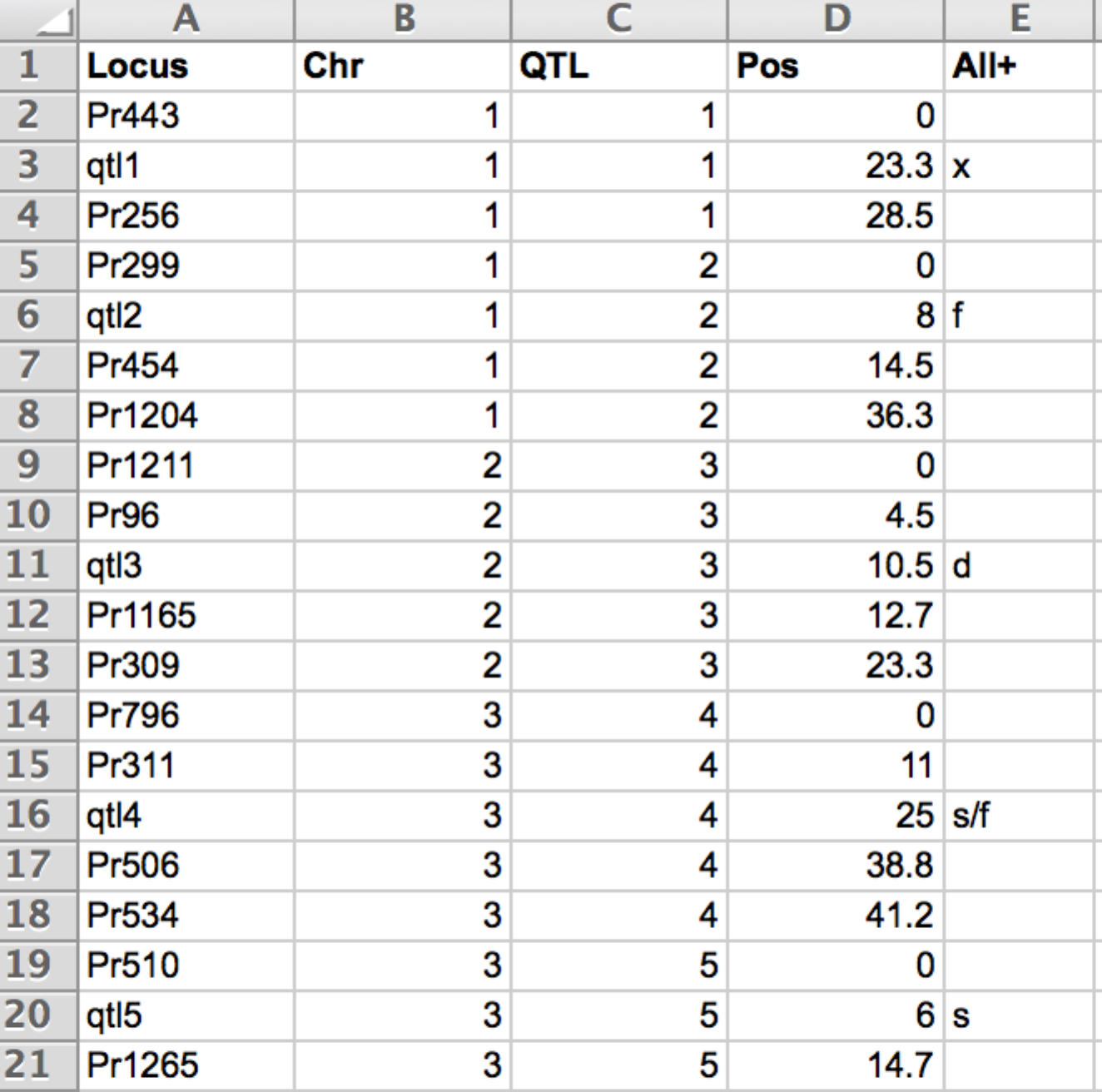
5 of the 11 QTL and flanking markers described by their map position (pos) in cM
Map File Columns
- Locus: Mame of markers and QTL. QTL name are prefaced with "qtl".
- Chr: Chromosome
- QTL: QTL numbered in ascending order
- Pos: (cM) For QTL, this is the estimated QTL position coming from the QTL detection results. Positions of the different loci must be obtained using the Haldanes mapping function (i.e. by assuming no interference).
- All+: Parental allele considered favourable for the QTL
Genotype/Pedigrees File (.dat)
The genotype/pedigree file is in plain-text tab-delimited format (no space between fields). Column headings A-F should not be changed even if the 2 optional columns, Cycle and Group, are left blank (or -). Column headings begining with G describe the 34 microsatalite markers. This format is very close to the input format of the Flapjack software.
Example File: ?blanc.dat
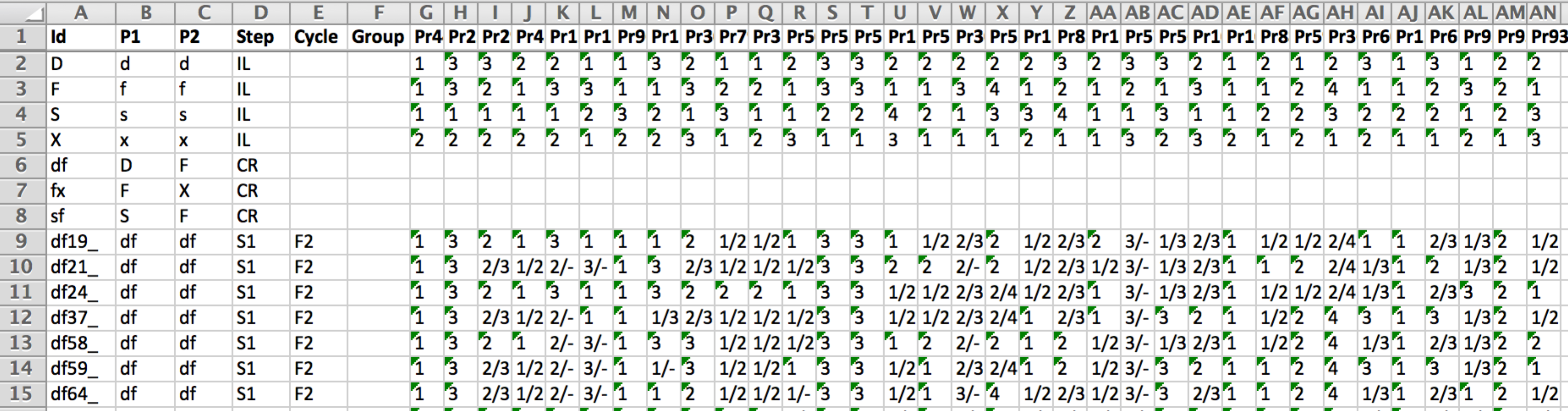
15 of the 279 genotyped lines: The first four rows are inbreed parental lines (IL) homozygous for all 34 markers. Rows 6-8 are heterozygous F1s, and are not genotyped. Rows 9-15 are genotyped F2 individuals. Markers are numerically coded; A=1, C=2, G=3, T=4.
Genotype/pedigree file column descriptions
- Id: The name of each genotyped individual. Individual IDs must unique and ranked according to generations (from oldest to most recent). The first individuals of this file must correspond to the founder parents of the program (here D,F,S, & X) that are assumed to be homozygous lines.
- P1 & P2: Correspond to the name(s) of the parent(s) of the individual (must exist as individuals in above in the file). The pedigree of the parental lines is assumed to be unknown. For a founder line P1 and P2 columns indicate the name given to the allele (e.g. A, B or any other character) coming from this line that will be traced through generations.
- Step: Corresponds to the pedigree relationship between the individual and its parent(s):
- CR: Cross indicates that the individual results from a cross between its two parents.
- Sn: Selfing indicates that the individual results from n generations of selfing of its parent (in this case the two parent Ids must be identical).
- RIL: Recombinant inbred lines, assumes that the individual results from an infinite number of selfing generations from an initial F1 hybrid. In this case, parent 1 and parent 2 must be identical and correspond to the F1 hybrid.
- DH: Double haploids assumes that the individual results from haplo-diploidisation from an initial F1 hybrid
- IL: Inbreed line indicates founder inbred status
- Cycle: Optional information regarding the generation in the program (e.g. F2, F4, C1, etc.).
- Group: Optional information regarding another classification criterion (e.g. subprograms, families, etc.).
- Mk1-Mkn: These are the genotyping results. The software can deal with SNPs, microsatellites, and any bi/multi allelic marker genotyping technique with either dominant or codominant scoring. The markers present in the genotypes/pedigree file must be ordered and match those in the map file (same number of markers). Homozygous genotypes for an allele (e.g. A) can be scored either as A or A/A. Heterozygotes are expected to be separated by a / (e.g. A/G). Heterozygous genotypes are assumed to be unphased (i.e. A/B equivalent to B/A). Missing data at marker loci are allowed and must be entered as - (or can be left blank and corrected later see below). For dominant markers, assuming A dominant vs. a recessive, genotypes presenting allele A must be coded A/-. Parental inbred lines should not contain missing data. Given the possibility to include non-genotyped individuals, this makes it possible to analyse most common marker-assisted selection schemes and mating designs.
Run OptiMAS
- Launch OptiMAS from the workbench or from the OptiMAS executable file located in the Tools folder.

- Select File > Import Data from the menu bar and browse for the genetic map file (.map) and genotypes/pedigree file (.dat) Set the location of the output directory. Results from each run will be stored within a new dated directory created automatically within this folder. Note that your output directory should not be in the Program Files folder or other specific directories with administrator privileges. Select Run button to analyse the data set. Close the Import Data window when the progress bar displays 100%.
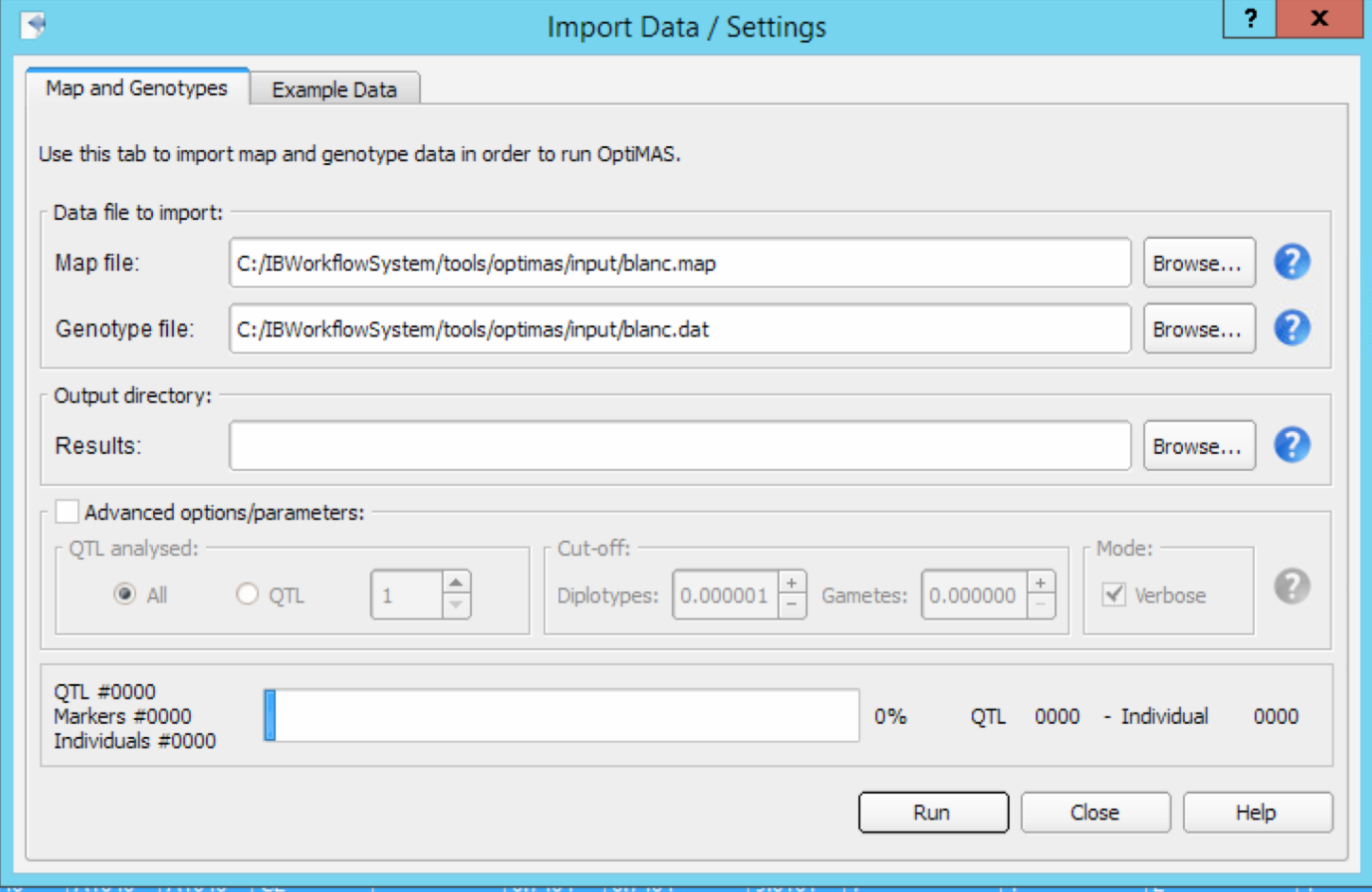
Advanced options/parameters
- QTL Analysed: By default all the QTL present in the input files will be analysed. You can also choose to select a specific QTL to run the analysis.
- Cut-off -Diplotypes: Genotypic probability below which a rare phased genotype (diplotype) is removed and not considered in subsequent computations (default value = 0.000001), to reduce memory and time needed for computation
- Cut-off-Gametes: Gametic probability (default value = 0.000000) that corresponds to the probability that the number of crossovers expected in the region between flanking markers exceeds a given value. Thus, unlikely haplotypes, or gametes, with number of crossovers over this value are removed and not considered in subsequent computations. Use of this option with values up to 0.01 is recommended in case many flanking markers per QTL lead to high computation time with default option.
- Verbose: Verbose mode creates two files per QTL position, reporting respectively gamete and diplotype probabilities for all individuals (default value = ON).
Correct Missing Genotype Data
A .cmap file has been created for import into the FillMD@Mks tool, which will fill in missing data with a placeholder and reconfigure the genotype/pedigree (.dat) file for reanalysis. A genotype/pedigree file may contain missing data due to genotyping errors. If a warning message appears after running OptiMAS, missing data may need to be replaced with a placeholder (-). Load input (.cmap and .dat) and set output directory, the path to the folder where the results will be stored. The .cmap file created at the end of a previous run of OptiMAS is used to re-run OptiMAS marker by marker and localize genotyping errors at individual marker position. The (.dat) file is the same genotype pedigree file used to run OptiMAS the first time. The output directory sets the destination of a new genotype/pedigree file (.dat) missing data is filled with a placeholder (-).
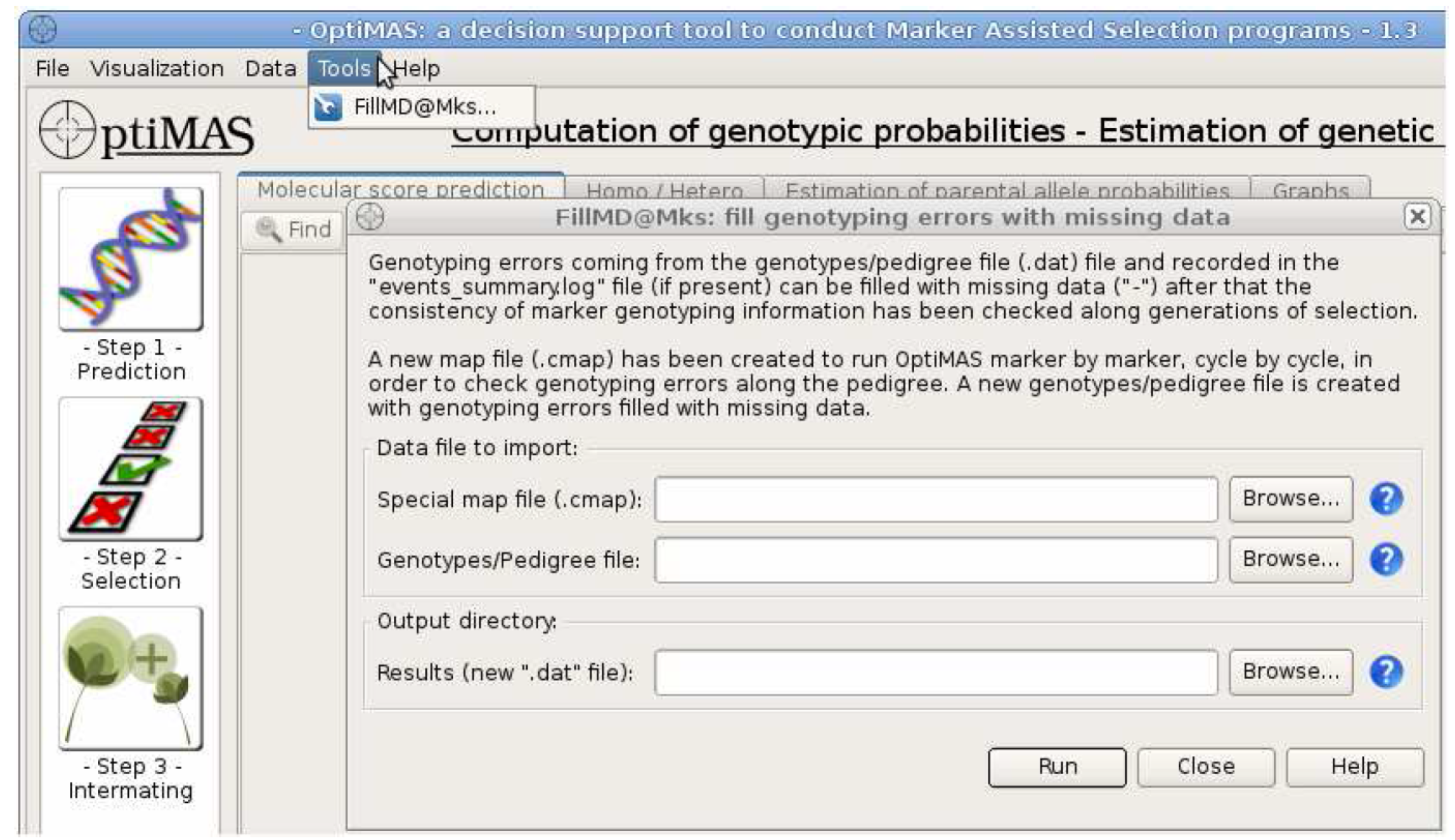
Note: It is also possible to directly display the results of previous analyses by selecting File > Reload data. You can also display results from the two examples data sets provided with the program, that are located in File > Example Data > Multiparental from the menu bar.
Prediction
Each QTL is given a score based on its probability of transmitting favourable alleles to gametic genotypes, or haplotypes. Genotyping data is phase ambiguous when markers are heterozygous, so OptiMAS calculates all possible phased diplotypes along with their probabilities, and then determines all of the possible gametes (haplotypes) and their probabilities at each QTL. OptiMAS summarizes the predicted genetic value of genotyped individuals into several metrics that inform selection.
- Access Prediction Functions by clicking Step 1 from the left menu.

Molecular Score Prediction Table
Genetic values and QTL scores are summarized in the Molecular Score (MS) Prediction window. Data can be sorted by MS or other criterion by double clicking the column heading.
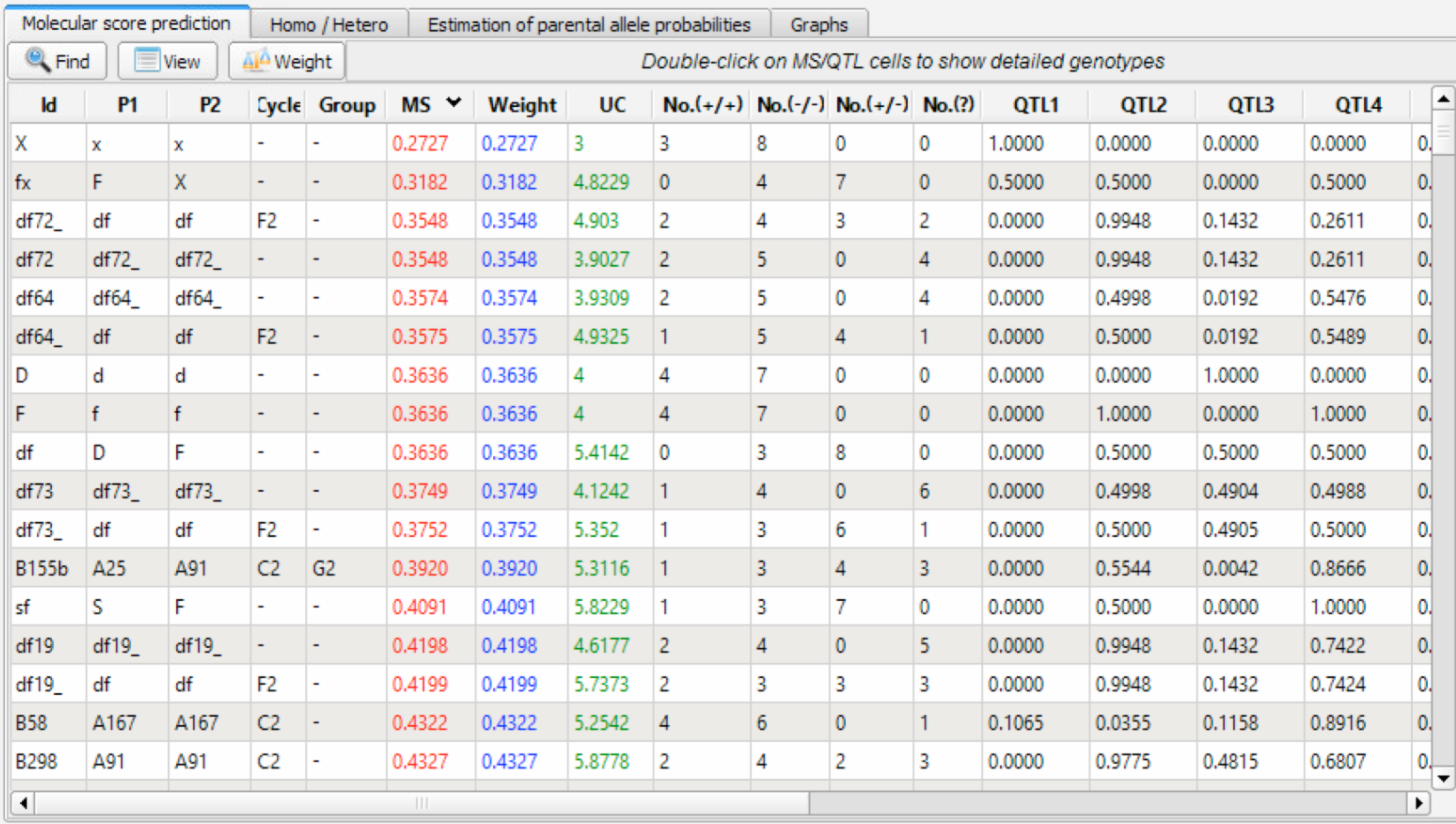
Molecular score prediction table sorted in ascending order by MS
Column Descriptions
- The first five columns correspond to the first five columns of the genotype/pedigree (.dat) file (see Genotype/Pedigree File Structure section 7.5.1)
- MS: Molecular Score is the additive probability of transmitting favourable alleles over all QTL. MS varies between 0, for an individual who does not carry any of the favourable alleles, to 1, for an individual which is homozygote for the favourable alleles. Individuals with a MS of 1 correspond perfectly to the target genotype.
- Weight: Breeder designated scaling of QTL importance in the breeding scheme (see QTL Weights section 7.7.5)
- UC: The utility criterion (UC) combines molecular score of an individual with the expected variance of gametic molecular scores possible from that individual. UC is based on the estimation of the expected number of favourable alleles carried by the superior 5% gametes produced by the individual. This criterion favours individuals with no fixed unfavourable alleles. This score ranges from 0 to the number of QTL. Note that present version of UC estimation assumes independence between QTL and should be considered as only indicative in the case of linked QTL. UC also assumes that the distribution of scores can be approximated by a normal distribution, which is not a valid assumption in case of small number of heterozygous QTL.
- No.(+/+): Number of QTL homozygous for favourable allele(s). A given QTL is considered homozygous for favourable allele(s) when the probability (+/+) exceeds a default threshold value of 0.75 (see Change Default p. 151).
- No.(-/-): Number of QTL homozygous for unfavourable allele(s). A given QTL is considered as homozygous for unfavourable allele(s) when prob (-/-) exceeds a default threshold value of 0.75 (See Change Default).
- No.(+/-): Number of QTL heterozygous with both favourable and unfavourable allele(s). A given QTL is considered to belong to this category when prob (+/-) exceeds a default threshold value of 0.75 (See Change Default).
- No.(?): Number of QTL defined as uncertain. Concerns QTL which are not attributed to any of the three previous categories.
- QTL: Individual molecular scores corresponding to each QTL
Molecular Score Details
- Display more detailed genotypes by double clicking on MS or QTL cells. This view summarizes and aggregates the information presented in the Homo/Hetero and Estimation of Parental Allele Probabilities tables.
- Double click the cell representing QTL 2 of individual B8 to reveal a yellow popup box.
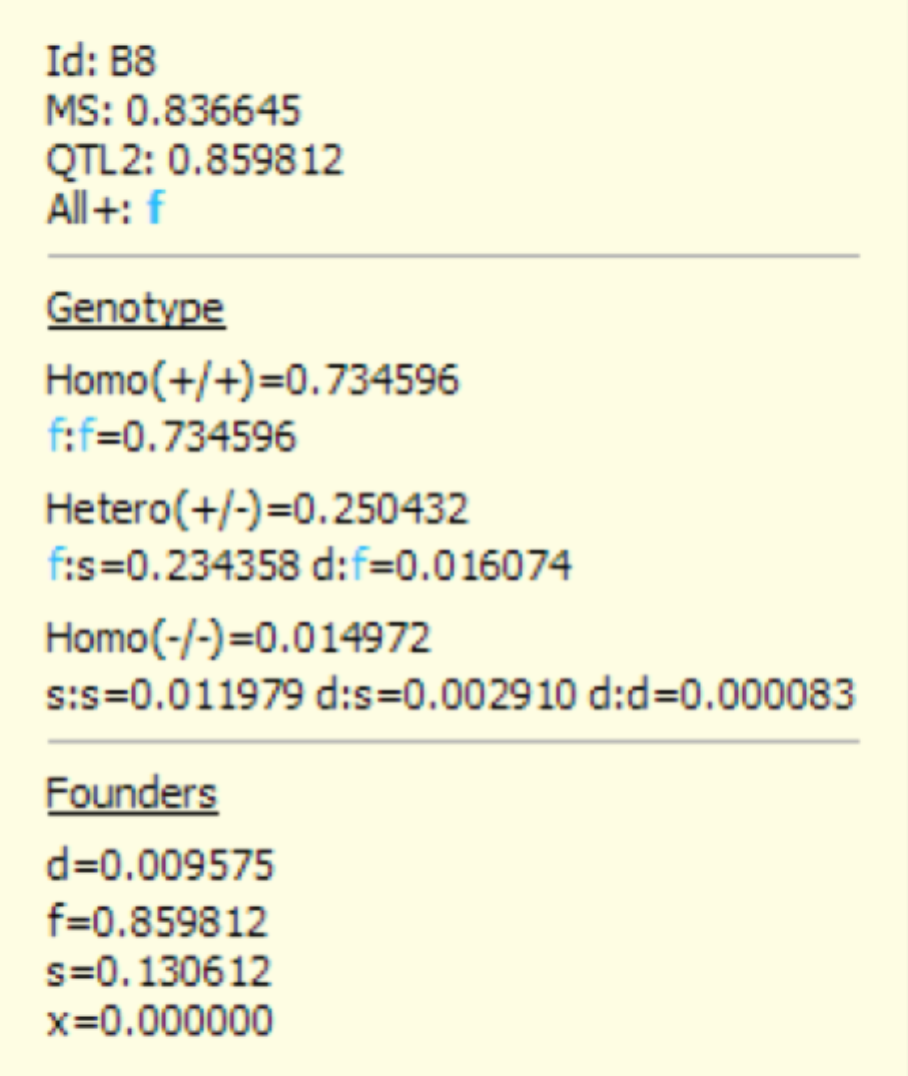
B8 has a molecular score of 0.8366451 at QTL 2, which has a probability of 0.734596 to be homozygous for the favourable allele f (i.e. Homo(+/+)). This score corresponds to the sum of the probabilities of the genotypes f:f=0.734596, f:s=0.234358 and d:f=0.0167074. QTL 2s MS of 0.995832 corresponds to the expected proportion of favourable allele(s) (i.e. Homo(+/+) + ½ Hetero(+/)). Founders, represented by the d,f,s, and x alleles, indicate the expected proportion of parental alleles.
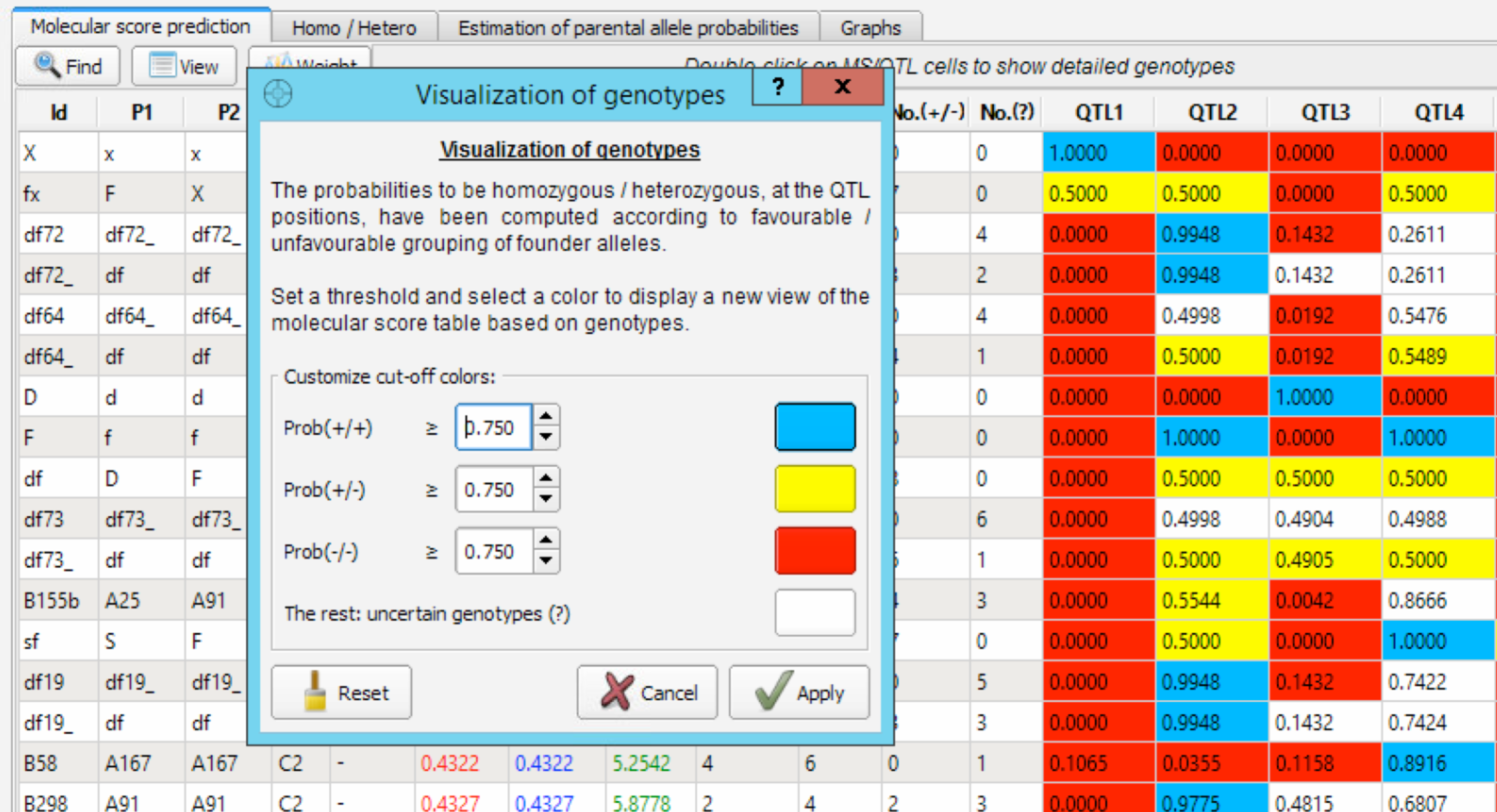
Visualize Genotypes
- Select and open the Visualization of Genotypes window.
The Visualization of Genotypes window creates a custom color-coded view of the molecular score table to more easily identify fixed QTL. The default value of 0.750 for cut-off colours can be altered from this window. When you apply a new set cut-off/colour parameters, the four corresponding columns [No.(+/+), No.(-/-), No(+/-), No.(?)] on the MS table are updated. Uncertain genotypes can be due to (1) recombination near the QTL position making one flanking marker heterozygous and the other being homozygous favourable or (2) due to missing data in the genotypes/pedigree (.dat) file.
Graphical Output
- Select graphs to visualize the tabular information. All the graphs can be exported as .png, .svg or .eps formats by selecting Save.
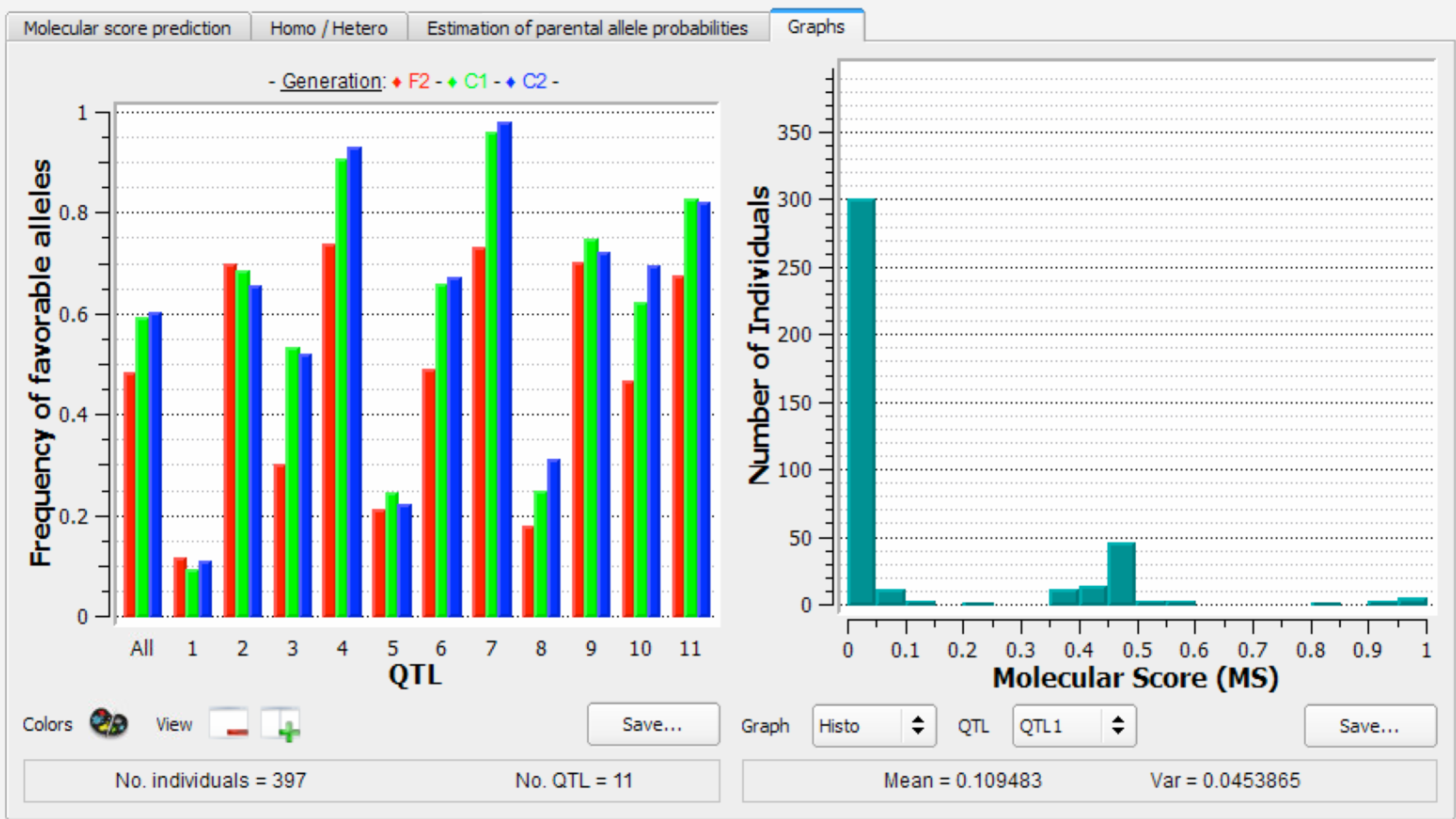
The left graph illustrates the frequency of favourable alleles at the different generations of selection. Note that no genetic gain is expected for the last generation (C2 in blue), because individuals have not yet been selected. The right graph is a histogram of the number of individuals with different molecular scores (MS) for QTL1 .
QTL Weights
- Examine the Weight column of the Molecular Score Prediction table.
The favorable allele at QTL 1 is in danger of being lost, because the 11 best individuals as defined by overall molecular score (MS) are fixed at 0 for this QTL.
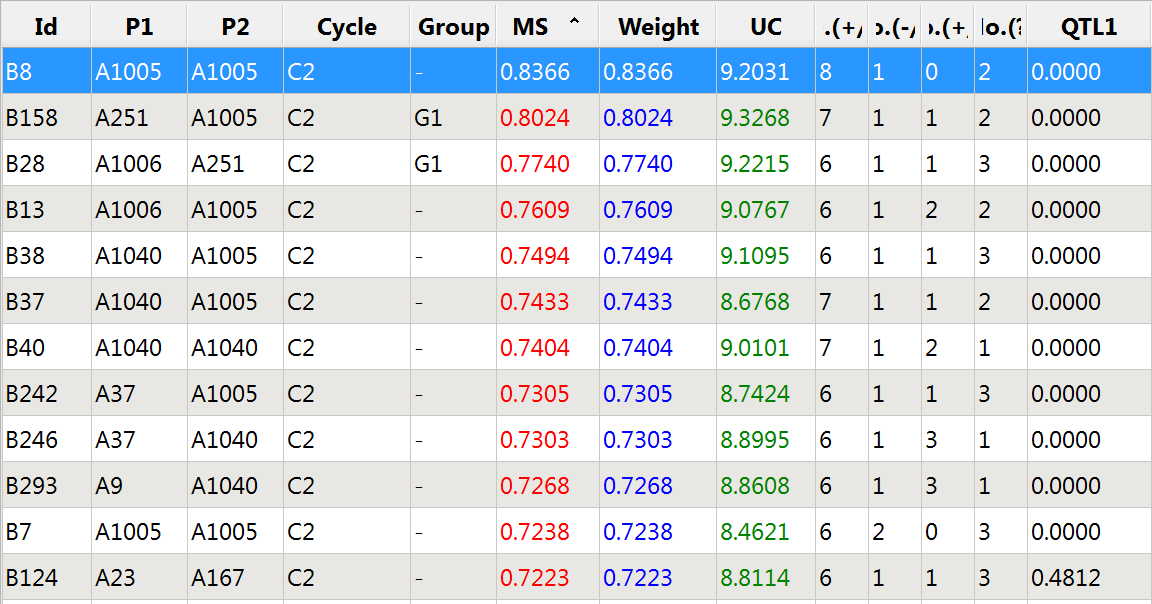
Top 12 germplasm based on molecular score (MS): Not weighted for QTL 1
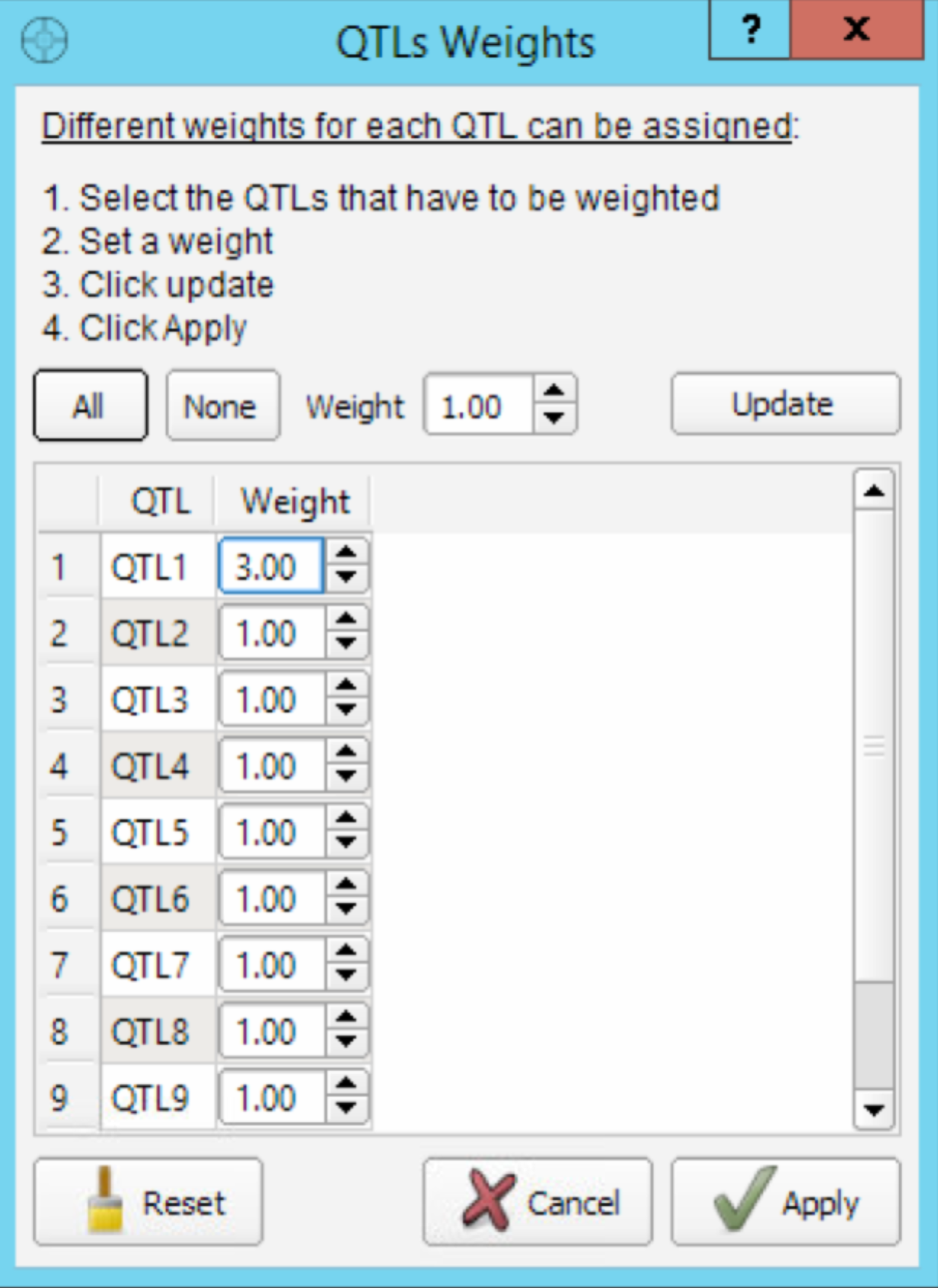
OptiMAS attributes the same weight to all QTL in the map file for molecular score (MS) estimation. However is possible to discard QTL and/or to attribute weights defined by the breeder by applying a Weight index to the MS calculation.
- Select the Weight button to open the QTL Weights dialog window. Assign QTL 1 a weight of 3 to produce a new classification of individual based on the new weighted MS.
Increasing the weight of QTL 1 allows germplasm to be sorted by weighted molecular score, and ensures that some of the top scoring germplasm can transmit the favorable allele.
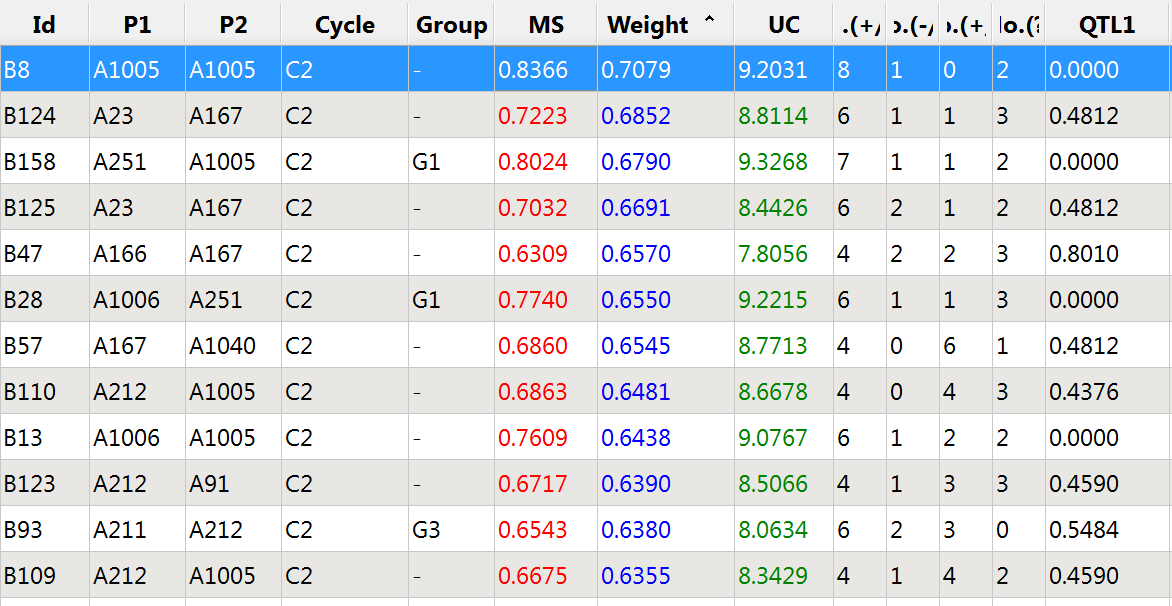
Top 12 germplasm based on weighed molecular score: QTL 1 weighted 3
Find Individuals on Molecular Score Table
The Find ID dialog box can be used to search and locate a specific individual in the panel.
- Select Find and enter 242.
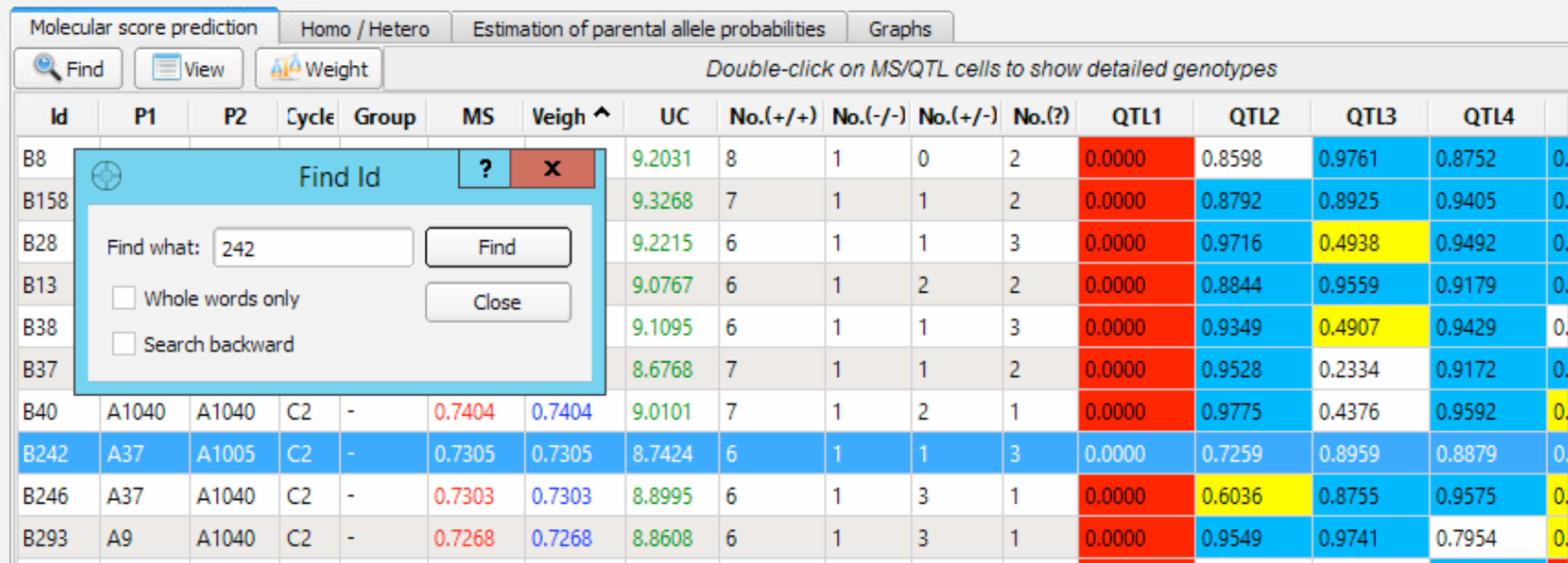
Filter Molecular Score Table
The QTL/Individuals dialog is used to enable or disable the display of QTL and/or individuals on the MS table.
- Press the View button to display the filter dialog. Select and check QTL and/or individuals to follow. Select OK to apply the corresponding filter and immediately refresh the MS table. This new view of the MS table can be useful if you are working with a large number of QTL and/or individuals and you want to focus on specific QTL/plants.
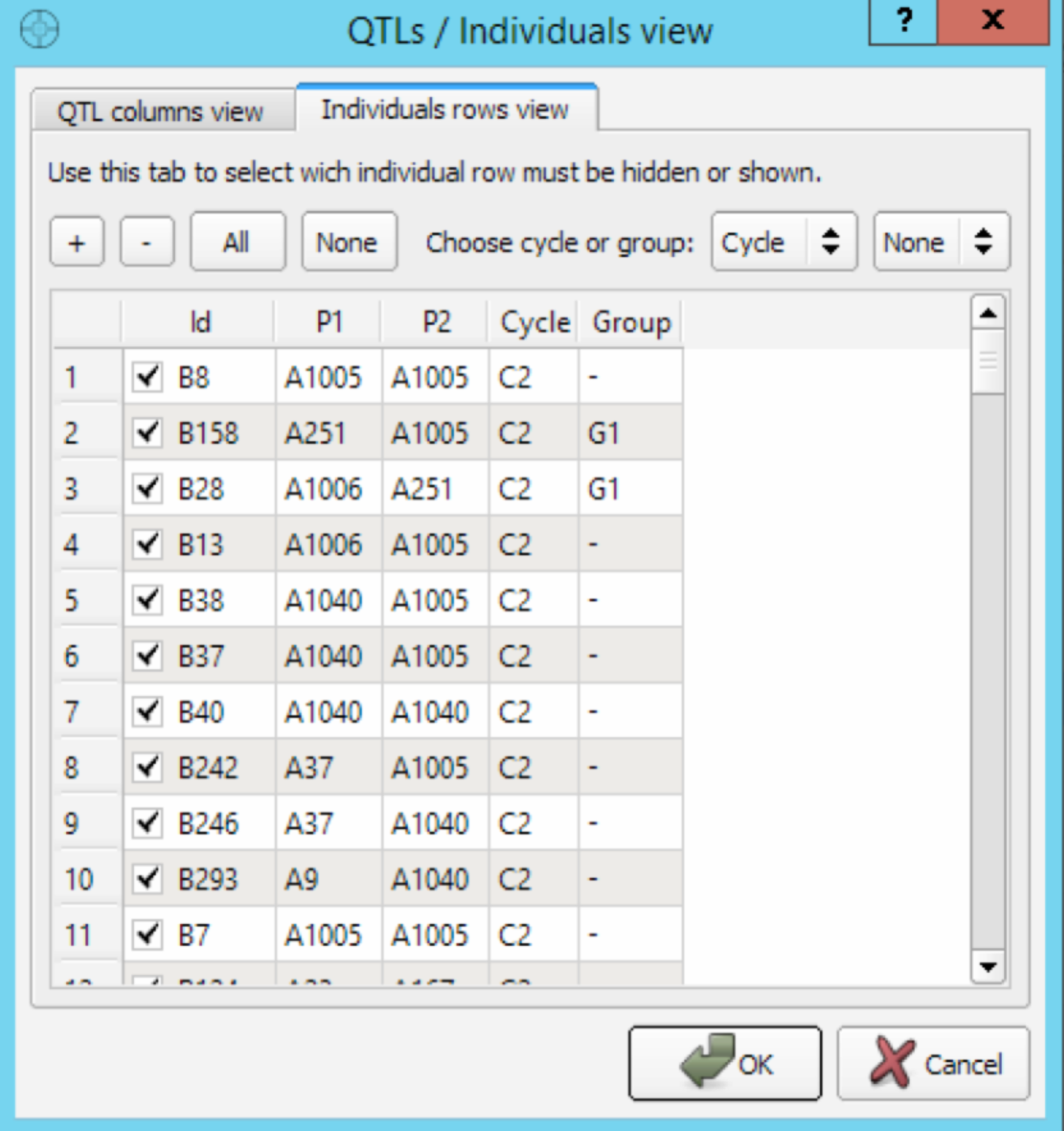
Selection
The selection features are accessed by choosing Selection. Genetic value predictions can be used in three different ways to inform selections:
- Manually select individuals based on own judgment
- Truncated selection based on molecular score (MS), weighted MS, or the utility criterion (UC).
- QTL complementation selection which aims to prevent the loss of rare favourable alleles
- Access Selection Functions by clicking Step 2 from the left menu.

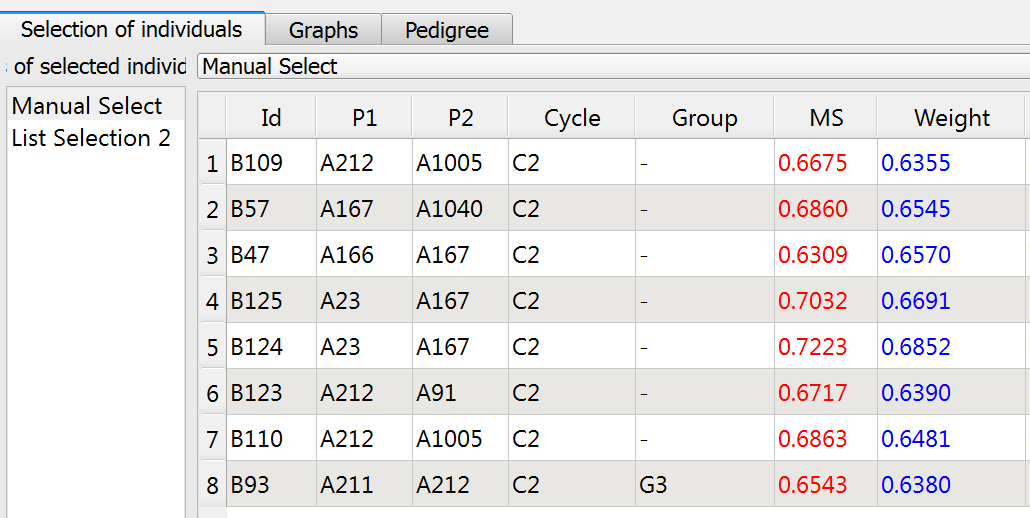
Manual Selection
- Create a custom list of germplasm with high weighted molecular scores, but without fixation at QTL1.
- Double click the following lines and add to to List Selection 1:
- B124
- B125
- B47
- B57
- B110
- B123
- B93
- B109
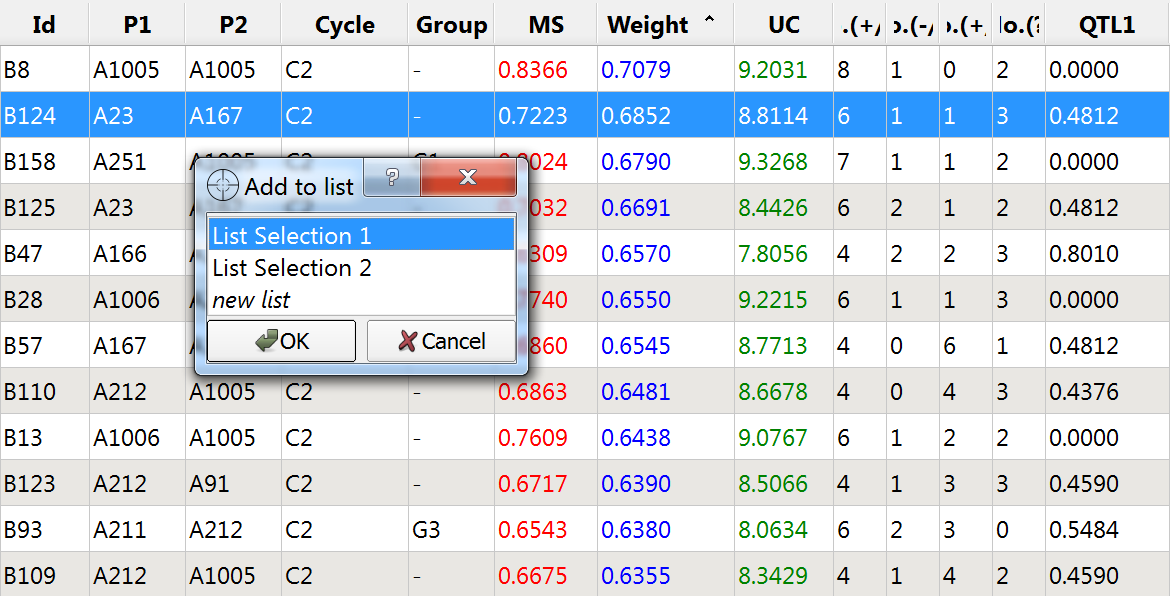
- Access List Selection 1 through the Selection Window. Double click on "List Section 1" and rename "Manual Selection"
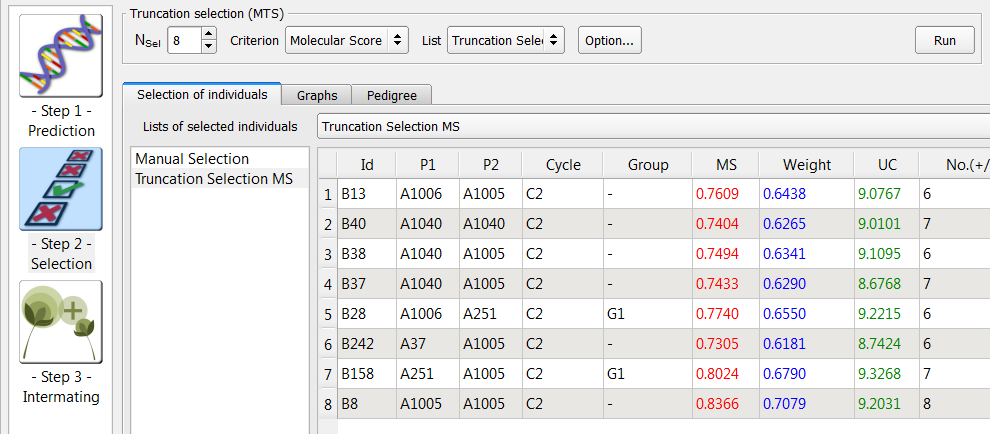
Truncation Selection
Truncated selections are automatically made bases on Molecular score (MS), weighted (MS) or utility criterion (UC). Set the truncation selction paramenters.
- Select 8 lines (Nsel) based on molecular score (MS). Rename "List Selection 2", "Truncation." Select the Truncation list and click run.
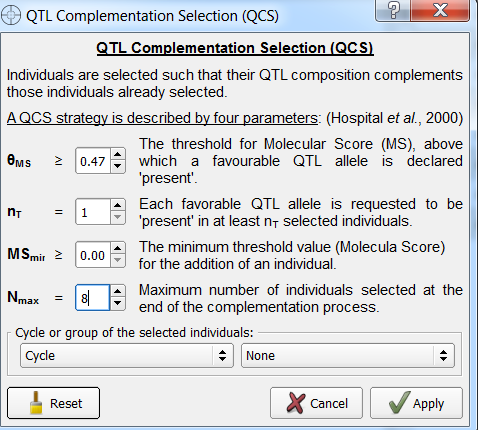
QTL Complementation Selection
QTL complementation selection aims to prevent the loss of rare favourable alleles, and is recommended when high numbers of QTL are being considered. QTL complementation adds individuals that complement a list previously created by truncation selection.
- From the Selection window add a new list titled, "QTL Complementation". Select this list for QTL complementation and click Option.

- Set Nmax to 8. Leave the other options in their default settings. Select Apply and Run QCS.
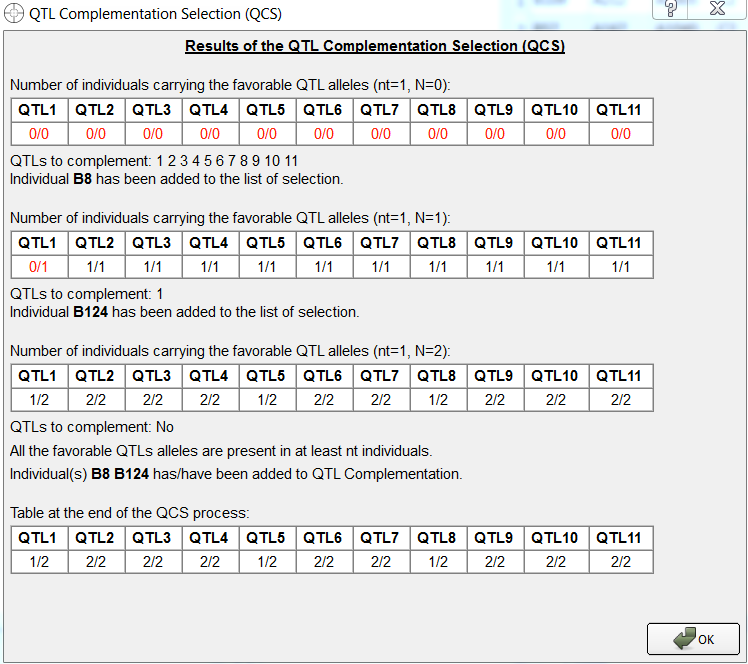
Two individuals, B8 and B124 have been identified by QTL complementation selection. B124 was not identified by truncation selection.
- Create a combined list of the 9 individuals selected by truncation selection and QTL complementation. Add a new list and name it "Selections." Drag and drop the unique lines to the new list.
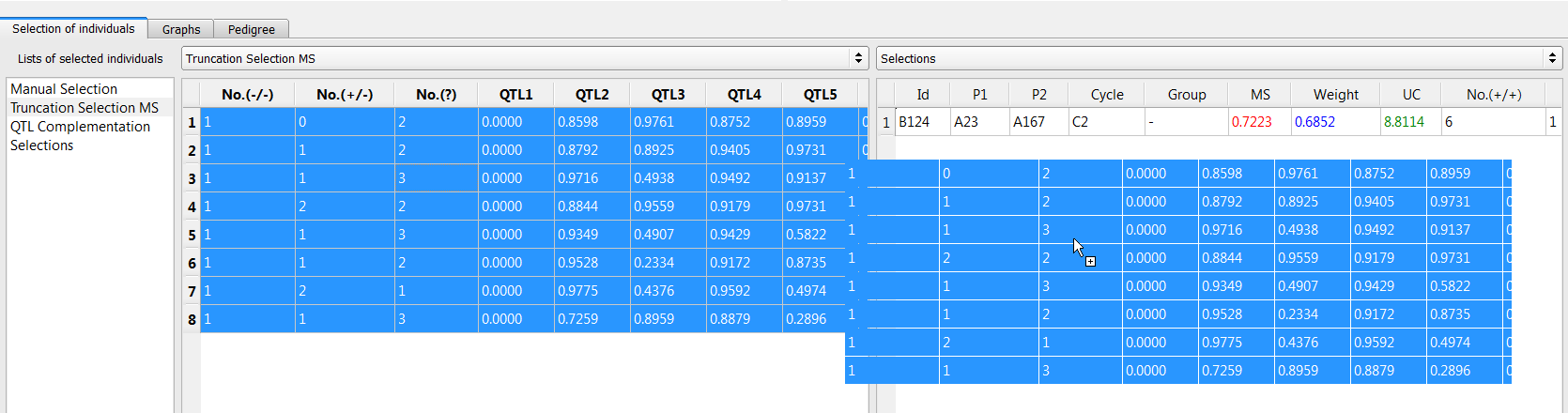
Graphs
- Select and open the Graphs tab allows for further comparisons of selection criteria.
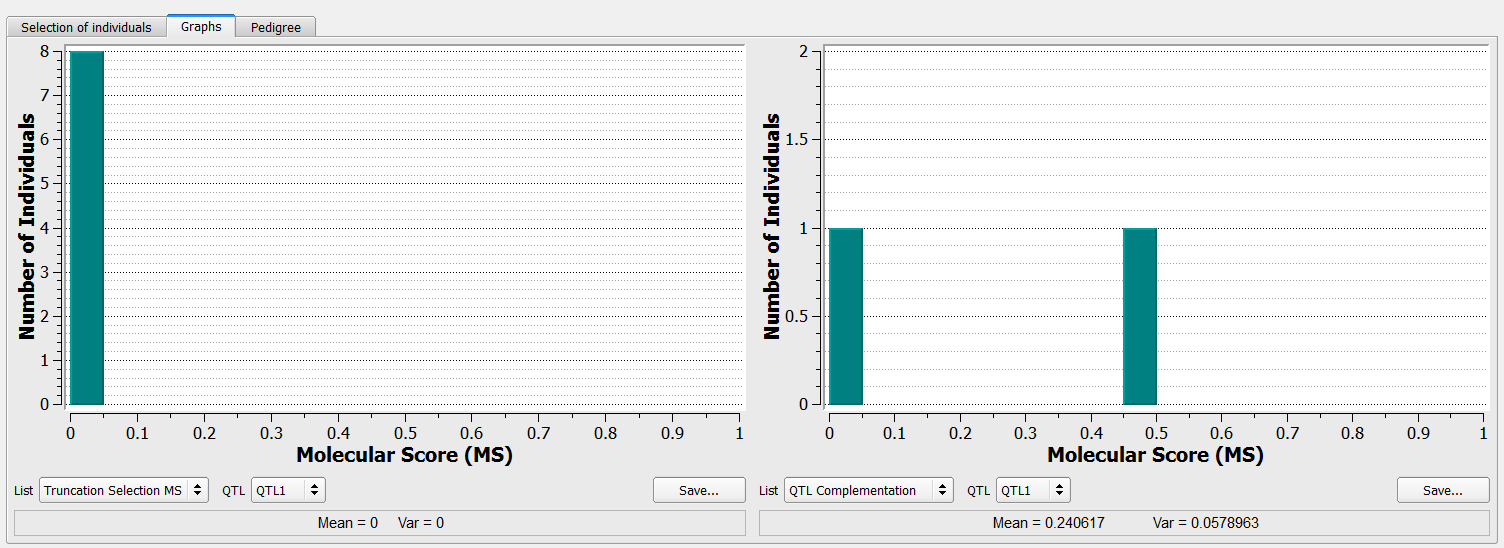
The left graph shows that all 8 individuals selected through truncation will carry the unfavourable allele at QTL1. The right graph on the includes the one individual, B124, added via the QTL complementation procedure that carries the favorable allele.
Selection Pedigrees
Pedigree illustration allows for easy visualization of the genetic contribution of selected individuals over generations.
- Choose the Selections list and indicate Alone to only display selected individuals. Click Generate.
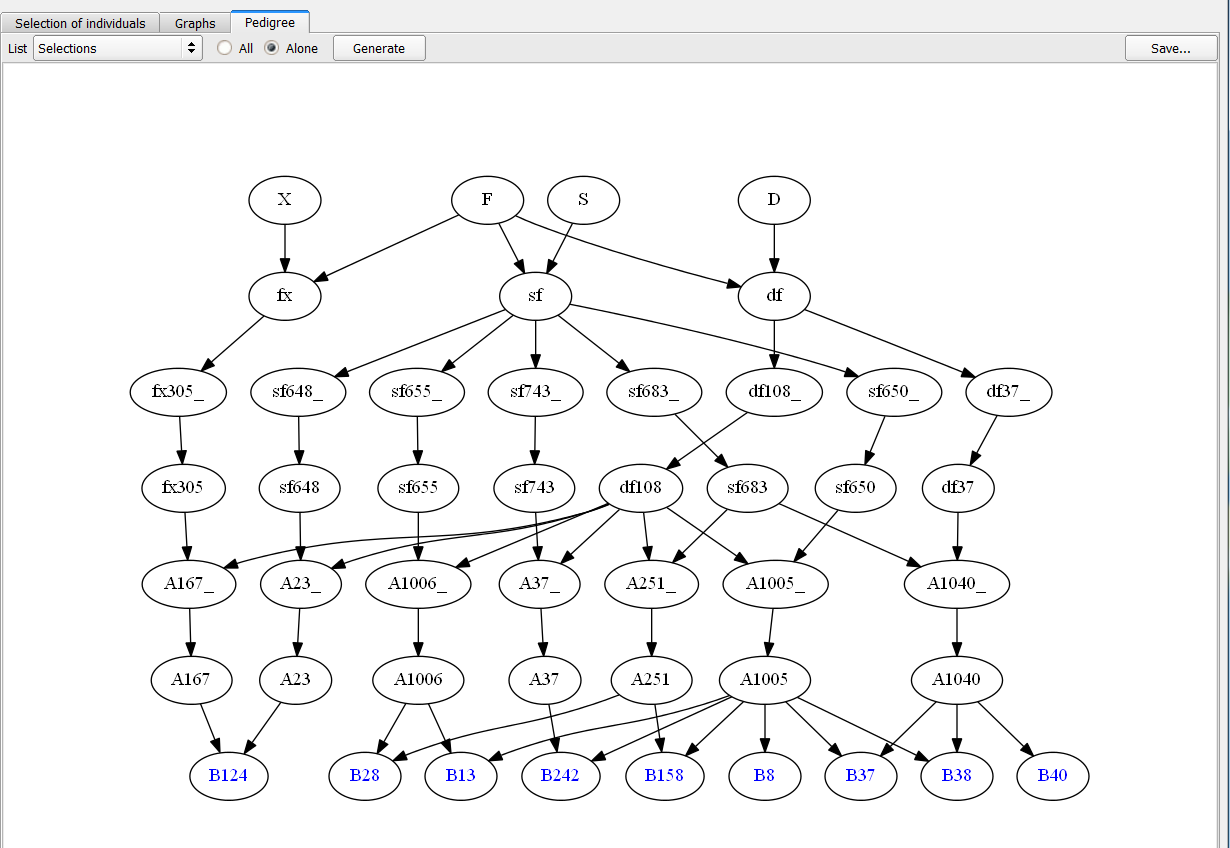
Pedigree of 9 Individuals Selected based on truncation and QTL complementation selection.
Intermating
Identify crosses to initiate the next round of marker-assisted selection using one of three design methods.
- The half-diallel complete method identifies crosses among all selected plants in a list
- The better half method identifies crosses within a list, while avoiding crosses between selected individuals with low genetic values
- Factorial Design Method: identifies crosses between two selection lists
Select the Intermating functions by selecting Step 3 from the left menu.

Half-Diallel Complete Method
The half-diallel complete method identifies crosses among all selected plants in a list.
Name an empty intermating list Half-diallel.
Choose the Selections list of 9 individuals selected based on truncation and QTL complementation selection.
Select the Complete method option and click Apply.
Assign the output to the Half Diallel list. Select Run.
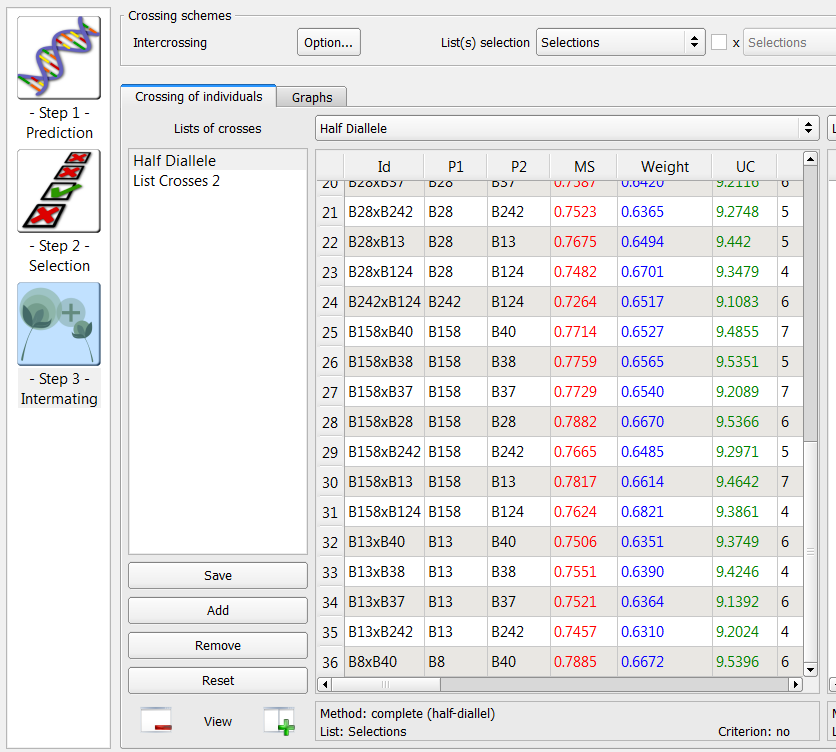
36 crosses generated by the half-diallele method on the list of 9 individuals selected based on truncation and QTL complementation selection
Better Half Method
The better half method identifies crosses within a list, while avoiding crosses between selected individuals with low genetic values. Name an empty intermating list Better Half.
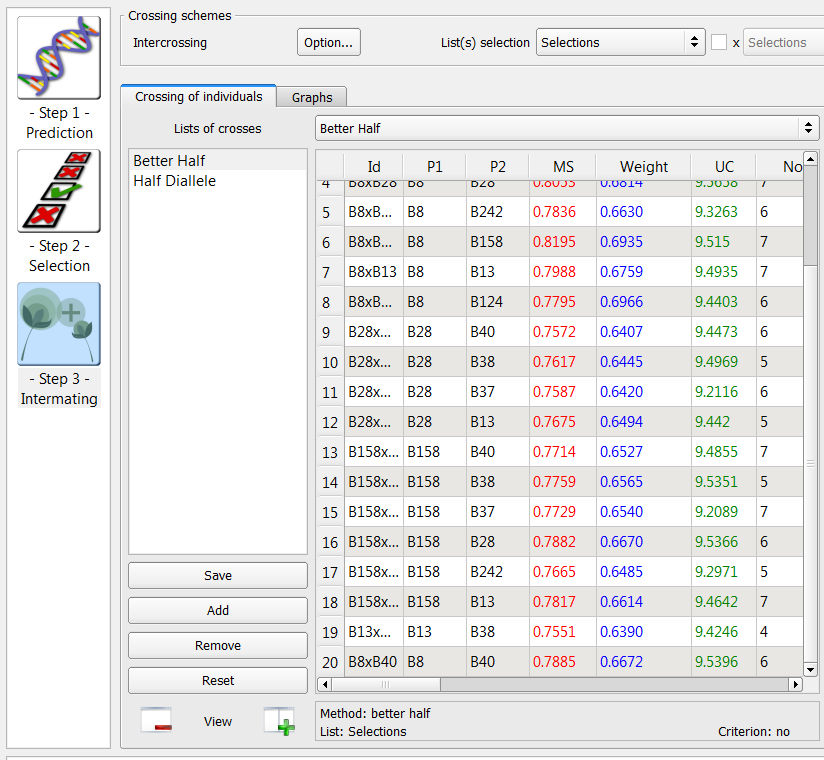
- Choose the Selections list of 9 individuals selected based on truncation and QTL complementation selection. Select the Better Half method option. Select Run.
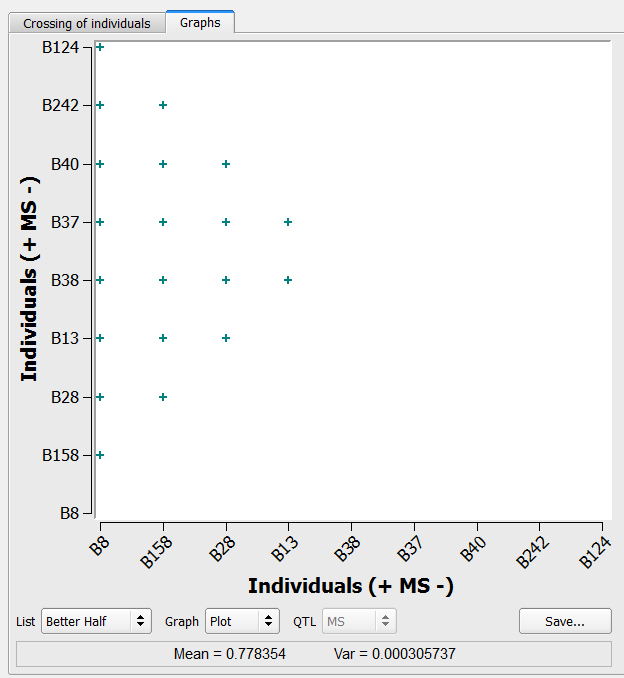
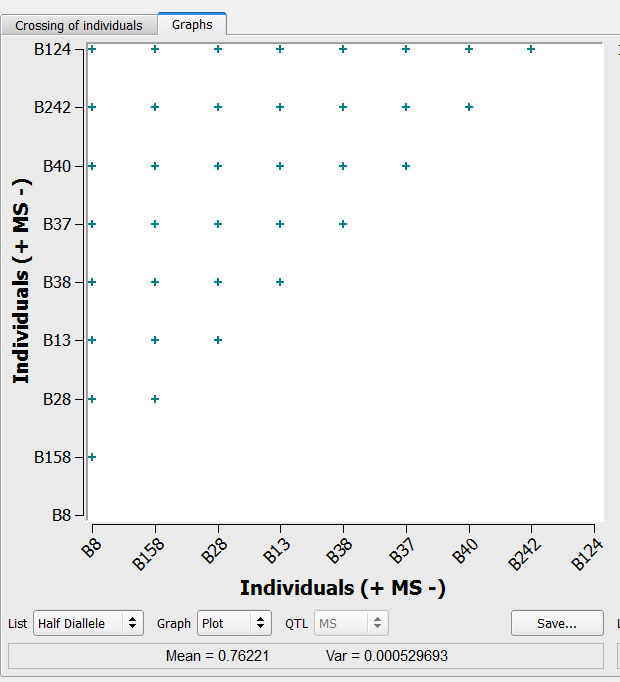
20 crosses generated by the better half method on the list of 9 individuals selected based on truncation and QTL complementation selection
Factorial Intermating Design
Intermating designs can be created using two selection lists by populating the additional list option.
- Cross the Manual selection (n=8) list with the Selection (n=9).
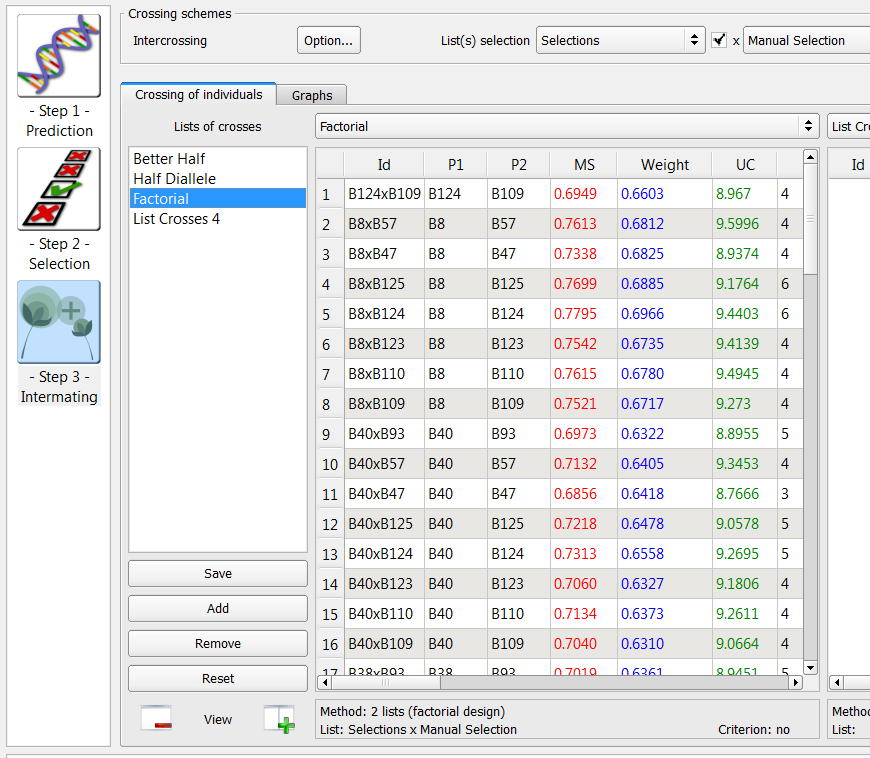
72 crosses generated by the factorial method on the list of 9 individuals selected based on truncation and QTL complementation selection and 8 individuals manually selected
References
Funding & Acknowledgements
The Integrated Breeding Platform (IBP) is jointly funded by: the Bill and Melinda Gates Foundation, the European Commission, United Kingdoms Department for International Development, CGIAR, the Swiss Agency for Development and Cooperation, and the CGIAR Fund Council. Coordinated by the Generation Challenge Program the Integrated Breeding Platform represents a diverse group of partners; including CGIAR Centers, national agricultural research institutes, and universities.
Maize mulitparental demonstration data were provided by Alain Charcosset. These data may have been adapted for training purposes. Any misrepresentation of the original data is the solely the responsibility of the IBP.

This work is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License.
